
Лаборатория компьютерного дизайна материалов
- DFT расчеты
- Молекулярное моделирование
- Атомистическое моделирование

Комната отдыха (405а НК)

Офис (405а НК)
Иван Круглов
Научный руководитель
Иван Горемыкин
Инженер
Георгий Бычков
Инженер
Людмила Березникова
Инженер-исследователь
Анастасия Зеленина
Младший научный сотрудник

Арслан Мазитов
Младший научный сотрудник
Александр Изьюров
Инженер-исследователь
Константин Кравцов
Лаборант-исследователь
Даниил Алексеев
Студент
Павел Маслов
Студент
Вячеслав Коротнев
Студент
Всего публикаций
52
Всего цитирований
2903
Цитирований на публикацию
55.83
Среднее число публикаций в год
4.73
Годы публикаций
2014-2024 (11 лет)
h-index
29
i10-index
42
m-index
2.64
o-index
95
g-index
52
w-index
9
Описание метрик
h-index
Учёный имеет индекс h, если h из его N статей цитируются как минимум h раз каждая, в то время как оставшиеся (N - h) статей цитируются не более чем h раз каждая.
i10-index
Число статей автора, получивших не менее 10 ссылок каждая.
m-index
m-индекс ученого численно равен отношению его h-индекса к количеству лет, прошедших с момента первой публикации.
o-index
Среднее геометрическое h-индекса и числа цитирований наиболее цитируемой статьи ученого.
g-index
Для данного множества статей, отсортированного в порядке убывания количества цитирований, которые получили эти статьи, g-индекс это наибольшее число, такое что g самых цитируемых статей получили (суммарно) не менее g2 цитирований.
w-index
Если w статей ученого имеют не менее 10w цитирований каждая и другие статьи меньше, чем 10(w+1) цитирований, то w-индекс исследователя равен w.
Топ-100
Области наук
Журналы












Цитирующие журналы






Цитируемые журналы





Издатели
|
2
4
6
8
10
|
|
|
Springer Nature
10 публикаций, 19.23%
|
|
|
American Chemical Society (ACS)
10 публикаций, 19.23%
|
|
|
Wiley
7 публикаций, 13.46%
|
|
|
American Physical Society (APS)
7 публикаций, 13.46%
|
|
|
Elsevier
6 публикаций, 11.54%
|
|
|
MDPI
3 публикации, 5.77%
|
|
|
Royal Society of Chemistry (RSC)
2 публикации, 3.85%
|
|
|
AIP Publishing
2 публикации, 3.85%
|
|
|
American Association for the Advancement of Science (AAAS)
1 публикация, 1.92%
|
|
|
Optica Publishing Group
1 публикация, 1.92%
|
|
|
IOP Publishing
1 публикация, 1.92%
|
|
|
Institute of Electrical and Electronics Engineers (IEEE)
1 публикация, 1.92%
|
|
|
2
4
6
8
10
|
Организации из публикаций











































































Страны из публикаций
|
5
10
15
20
25
30
35
40
45
50
|
|
|
Россия
|
Россия, 48, 92.31%
Россия
48 публикаций, 92.31%
|
|
Китай
|
Китай, 23, 44.23%
Китай
23 публикации, 44.23%
|
|
США
|
США, 12, 23.08%
США
12 публикаций, 23.08%
|
|
Армения
|
Армения, 7, 13.46%
Армения
7 публикаций, 13.46%
|
|
Великобритания
|
Великобритания, 7, 13.46%
Великобритания
7 публикаций, 13.46%
|
|
ОАЭ
|
ОАЭ, 7, 13.46%
ОАЭ
7 публикаций, 13.46%
|
|
Сингапур
|
Сингапур, 7, 13.46%
Сингапур
7 публикаций, 13.46%
|
|
Германия
|
Германия, 5, 9.62%
Германия
5 публикаций, 9.62%
|
|
Испания
|
Испания, 4, 7.69%
Испания
4 публикации, 7.69%
|
|
Швейцария
|
Швейцария, 4, 7.69%
Швейцария
4 публикации, 7.69%
|
|
Япония
|
Япония, 4, 7.69%
Япония
4 публикации, 7.69%
|
|
Страна не определена
|
Страна не определена, 3, 5.77%
Страна не определена
3 публикации, 5.77%
|
|
Франция
|
Франция, 2, 3.85%
Франция
2 публикации, 3.85%
|
|
Египет
|
Египет, 2, 3.85%
Египет
2 публикации, 3.85%
|
|
Польша
|
Польша, 2, 3.85%
Польша
2 публикации, 3.85%
|
|
Бельгия
|
Бельгия, 1, 1.92%
Бельгия
1 публикация, 1.92%
|
|
Бразилия
|
Бразилия, 1, 1.92%
Бразилия
1 публикация, 1.92%
|
|
Израиль
|
Израиль, 1, 1.92%
Израиль
1 публикация, 1.92%
|
|
Индия
|
Индия, 1, 1.92%
Индия
1 публикация, 1.92%
|
|
Италия
|
Италия, 1, 1.92%
Италия
1 публикация, 1.92%
|
|
Норвегия
|
Норвегия, 1, 1.92%
Норвегия
1 публикация, 1.92%
|
|
Швеция
|
Швеция, 1, 1.92%
Швеция
1 публикация, 1.92%
|
|
5
10
15
20
25
30
35
40
45
50
|
Цитирующие организации



































































Цитирующие страны
- Мы не учитываем публикации, у которых нет DOI.
- Статистика пересчитывается раз в сутки.
Направления исследований
Применение методов машинного обучения для поиска новых оптических материалов
+
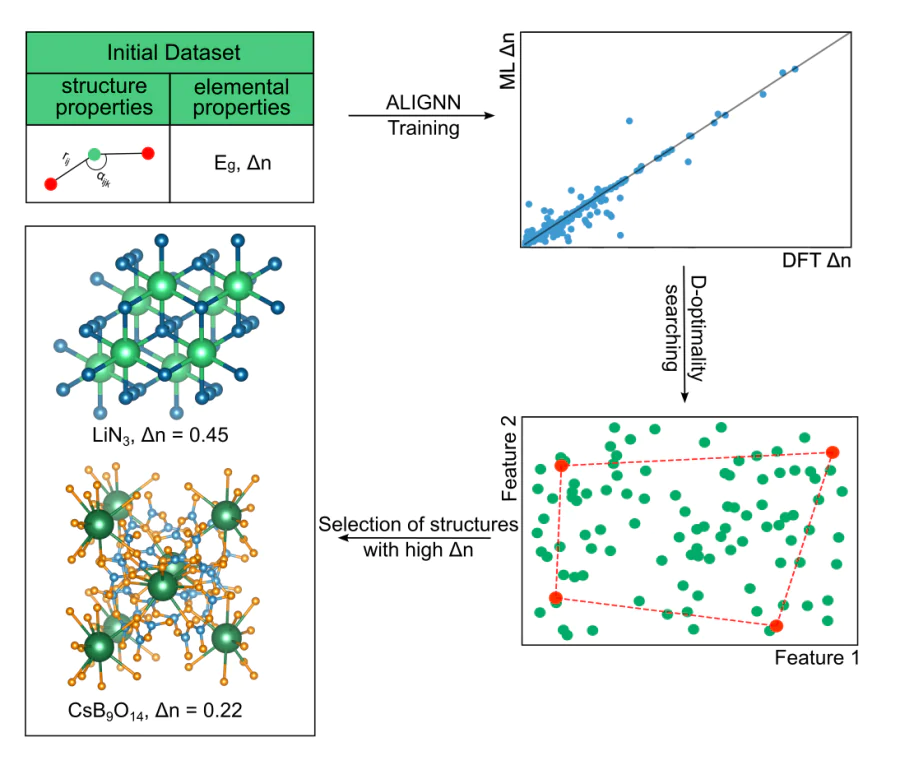
Одной из актуальных задач современного материаловедения является поиск оптических функциональных материалов с уникальными свойствами, такими как двулучепреломление и генерация второй гармоники, характеризующими их оптическую анизотропию. Эти свойства интересны благодаря их применению в самых разных областях науки и техники: поляризационных устройствах, оптической связи, фотолитографии и твердотельных лазерах. В рамках данного направления рассматривается возможность применения геометрических представлений кристаллической структуры и методов машинного обучения для выбора материалов с нужными характеристиками оптической анизотропии. Машинное обучение уже активно используется для предсказания различных свойств материалов на основе их структуры и химического состава, а некоторые из предложенных методов позволяют избежать дорогостоящих экспериментов. В нашей работе для прогнозирования оптических коэффициентов, характеризующих оптическую анизотропию материала, применяются графовые нейронные сети, показывающие свою эффективность в области вычислительного материаловедения. Полученные в ходе исследования результаты подтверждаются расчетами с использованием теории функционала плотности (DFT), а также верифицируются в эксперименте.
Высокотемпературная химия в углеродсодержащих материалах
+
Углерод способен порождать немыслимое количество аллотропных модификаций (по большей части метастабильных, но устойчивых), а также является каркасом всей органической химии. Для описания столь сложного элемента методами атомистического моделирования требуется комбинация самых продвинутых потенциалов (например, ReaxFF или машиннообучаемых потенциалов) и методов DFT.
С помощью такого инструментария мы:
объяснили, что происходит с оксидом графена при ультрабыстром ларезном нагреве и как это может помочь в оптимизации методик получения графена [1] (пресс-релиз - https://naked-science.ru/article/column/zakalka-lazernym-izlucheniem-uluchshila). По задачам, связанным с оксидом графена, в лаборатории выполняется грант РНФ 23−23−442 (рук. Орехов Н.Д.);
моделируем процессы пиролиза углеводородов и сажеобразования [2]. Этой теме был посвящен РНФ 20−79−245 (рук. Орехов Н.Д.);
изучаем фазовые переходы в чистом углероде в области экстремальных температур [3].
[1] (IF = 11.307) N Orekhov, et al «Mechanism of graphene oxide laser reduction at ambient conditions: experimental and ReaxFF study» // Carbon (2022).
[2] (IF=5.767) D Potapov, N Orekhov // «Mechanisms of soot thermal decomposition: Reactive molecular dynamics study» Combustion and Flame (2023).
[3] (IF = 11.307) N Orekhov, M Logunov // «Atomistic structure and anomalous heat capacity of low-density liquid carbon: Molecular dynamics study with machine learning potential», Carbon (2022).
Исследование магнетизма сильно коррелированных систем
+
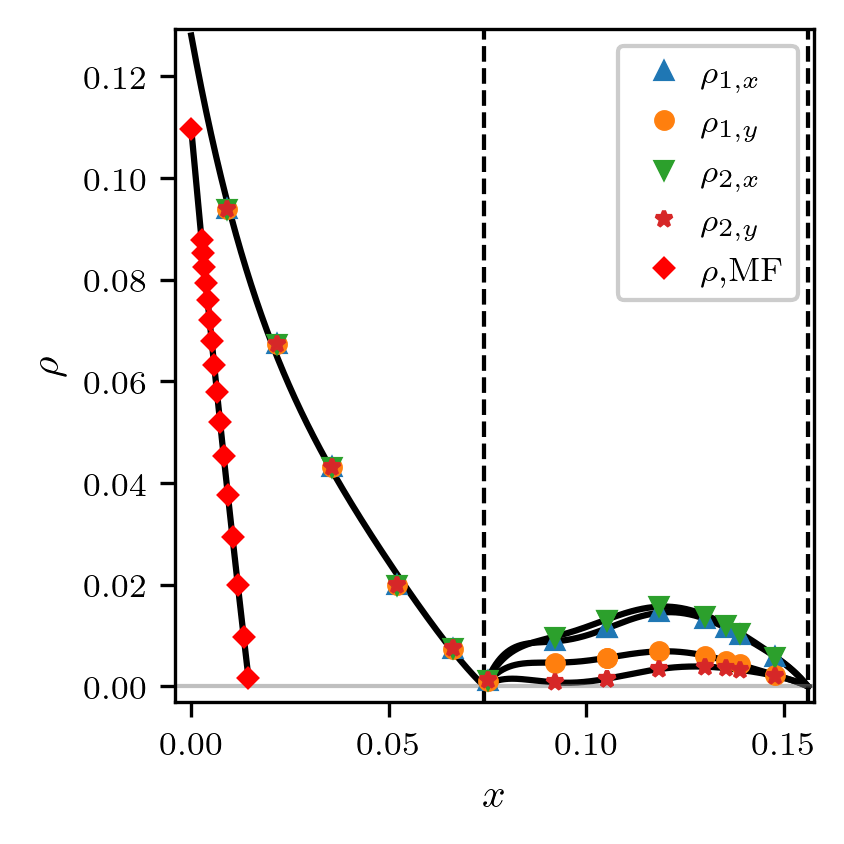
Электронные сильно коррелированные системы известны тем, что могут находиться в разнообразных фазовых состояниях, а их физические свойства определяются различными по своей природе и сравнимыми по энергетическому масштабу корреляциями. Магнитные корреляции в таких системах оказывают существенное влияние на свойства даже парамагнитных состояний вещества. Для исследования свойств сильно коррелированных систем с сильными магнитными флуктуациями нами применяются численные подходы, основанные на Динамической Теории Среднего Поля (DMFT). На данный момент одним из основных направлений нашей деятельности является исследование влияния геометрической фрустрации на сильно коррелированный магнетизм.
Первопринципная рамановская спектроскопия для надежной характеризации фотонных и оптоэлектронных материалов
+
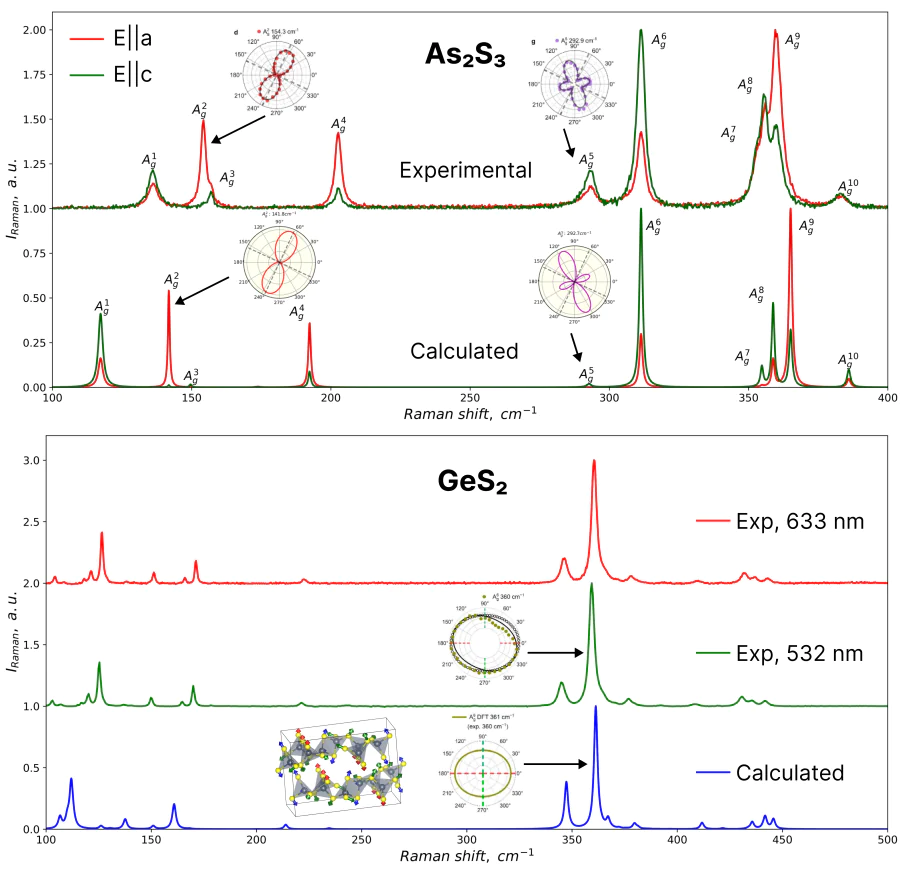
Ван-дер-Ваальсовы материалы привлекают внимание в науке и технике из-за их анизотропной кристаллической структуры и, соответственно, анизотропных свойств, таких как: двулучепреломление, модуль Юнга, тепло- и электропроводность и т. д. Они отрывают возможности для создания новых электронных, фотонных, оптоэлектронных, термоэлектрических и многих других устройств с выдающимися характеристиками.
Так, например, трисульфид мышьяка As2S3 и дисульфид германия GeS2 — уникальные ван-дер-Ваальсовы кристаллы с высокими измеренными коэффициентами преломления и двулучепреломлением. Но за шаг до того, как соединение будет внедрено в оптическое устройство, следует проверить его чистоту, поскольку примеси существенно влияют на оптические свойства твердых тел. Также важно знать положение кристаллографических осей материала с анизотропными свойствами для интеграции в устройство. Одним из способов подтвердить кристаллическую структуру и установить ориентацию исследуемого неметаллического вещества является использование рамановской спектроскопии. Это простой в использовании (по сравнению с рентгенографией), неинвазивный и, во многих случаях, неразрушающий метод (по сравнению с химическими методами).
Поэтому важной задачей является теоретическое предсказание спектра комбинационного рассеяния света не загрязнённых кристаллов, чтобы оценивать степень их чистоты в реальности для дальнейших экспериментов и применений. Вдохновленные этой проблемой, мы уже продемонстрировали на примере As2S3 валидность нашего подхода в работе [1] и скоро опубликуем работу, где отлично воспроизвелись спектры для GeS2, учитывая его очень сложную кристаллическую структуру. Кроме того, наши вычисленные полярные диаграммы для Раман-активных мод отлично согласуются с измеренными.
Таким образом, наш метод открывает большие перспективы для определения кристаллографических осей и проверки чистоты полупроводниковых материалов путем сравнения спектров Ab Initio с экспериментальными.
1. Slavich, A. S., Ermolaev, G. A., Tatmyshevskiy, M. K., Toksumakov, A. N., Matveeva, O. G., Grudinin, D. V., Voronin K.V., Mazitov A., Kravtsov K. V. , Syuy A. V., Tsymbarenko D. M., Mironov M. S., Novikov S. M., Kruglov I., Ghazaryan D. A., Vyshnevyy A. A., Arsenin A. V., Volkov V. S., Novoselov, K. S. (2024). Exploring van der Waals materials with high anisotropy: geometrical and optical approaches. Light: Science & Applications, 13, 68.
Публикации и патенты
Найдено
Ничего не найдено, попробуйте изменить настройки фильтра.

2024
—
2027
| Круглов Иван Александрович
Адрес лаборатории
Московская обл., г. Долгопрудный, Научный переулок, 4к1
Необходимо авторизоваться.